ETX101 for Dravet Syndrome

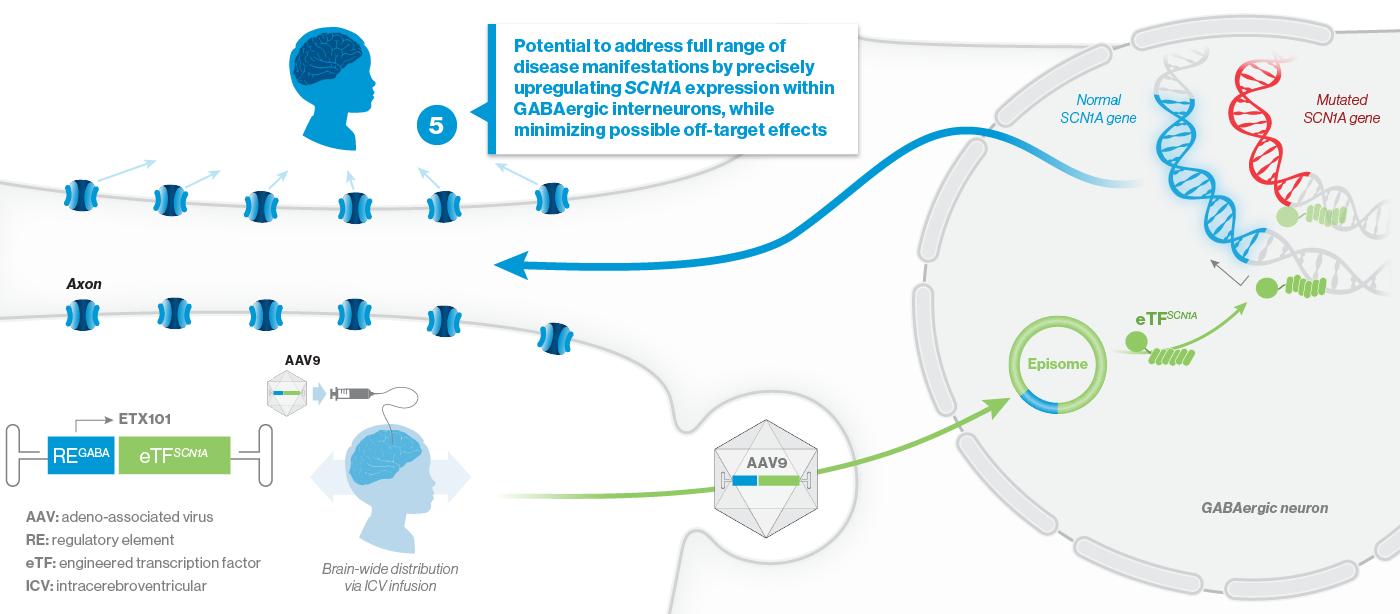
Encoded is developing ETX101, a potential one-time, disease-modifying gene regulation therapy for SCN1A+ Dravet syndrome. ETX101, Encoded’s lead program, is specifically designed to address the underlying cause of Dravet syndrome, the most common developmental and epileptic encephalopathy. ETX101 is a cell-selective gene therapy in development to potentially address the full range of seizure, cognitive, behavioral, developmental and motor manifestations of Dravet syndrome.
Dravet Syndrome Overview
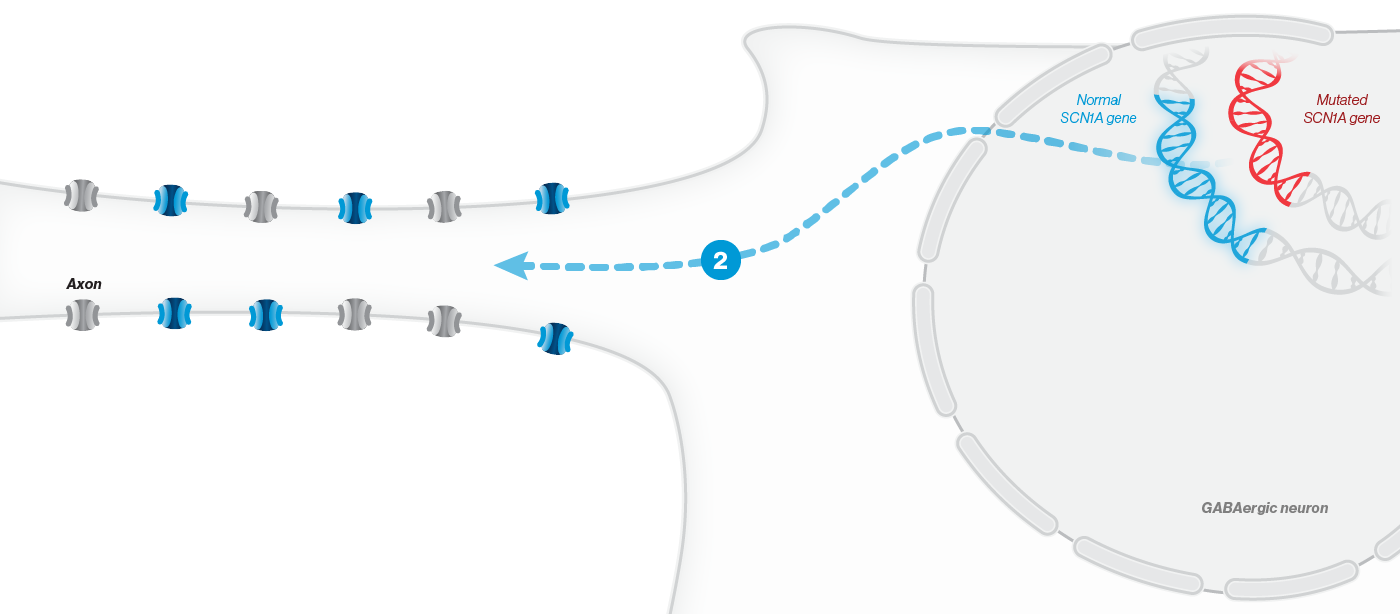
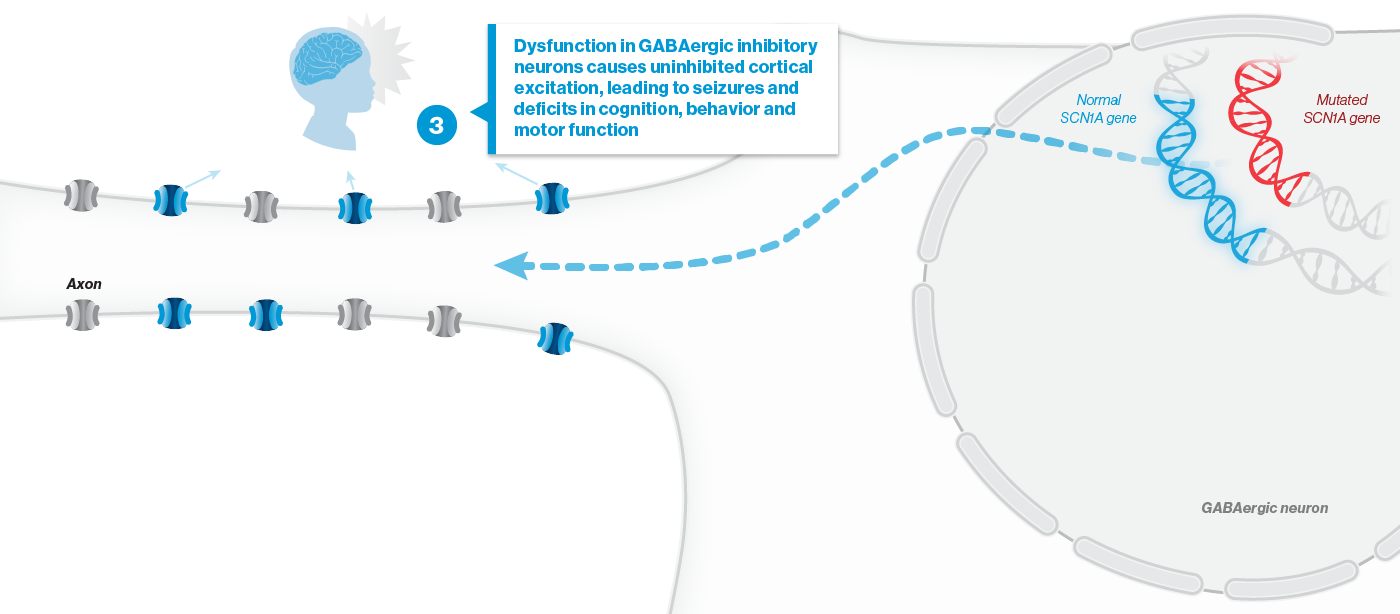
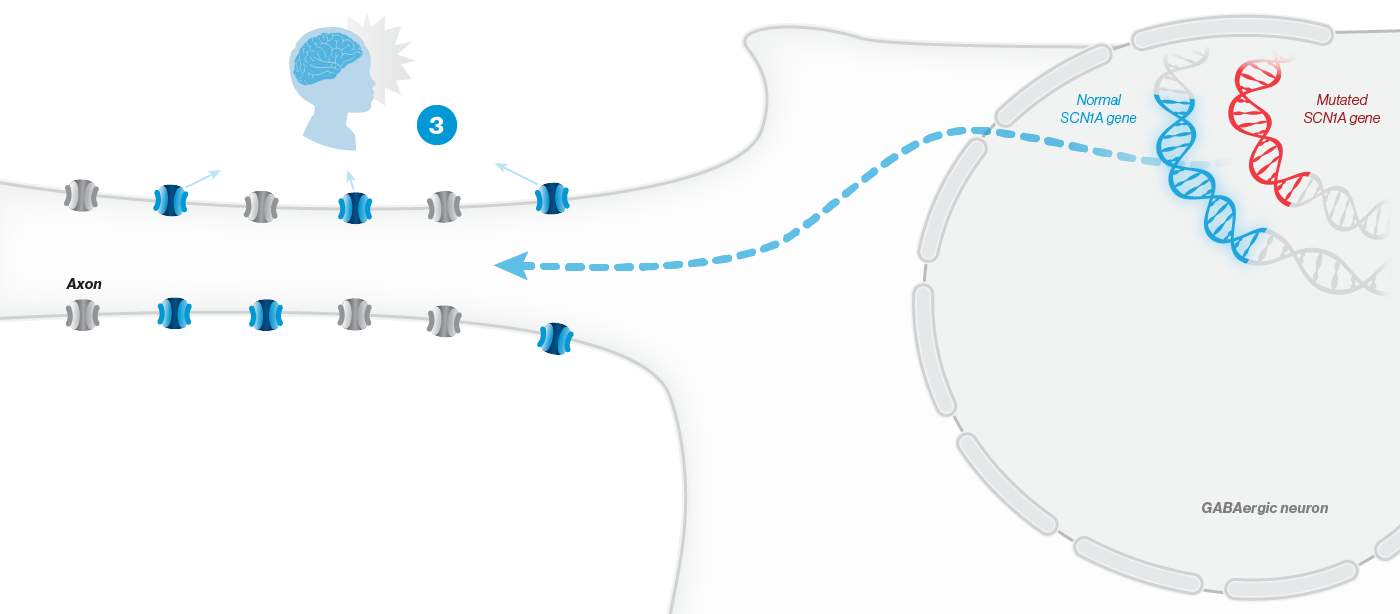
A mutation in the SCN1A gene causes about 90% of Dravet syndrome cases. Dravet syndrome is a disease of haploinsufficiency, meaning that only one of the two copies of the SCN1A gene works properly, leading to production of only half of the necessary sodium channels, NaV1.1, in neurons.
Devastating Disease in Pediatric Population
- Seizures typically present in the first year of life
Severe Disease with Impact on Mortality
and Neurodevelopment
- 1 in 5 children die before adulthood
- Significant developmental delay and cognitive disability
Large Unmet Need
- 1 : 15,500 incidence rate
- 35,000 patients in the US, EU3 (France, Germany, Italy), Canada and Japan
No Disease-Modifying Therapies
- Approved therapies reduce seizure burden but have limited impact on neurodevelopment
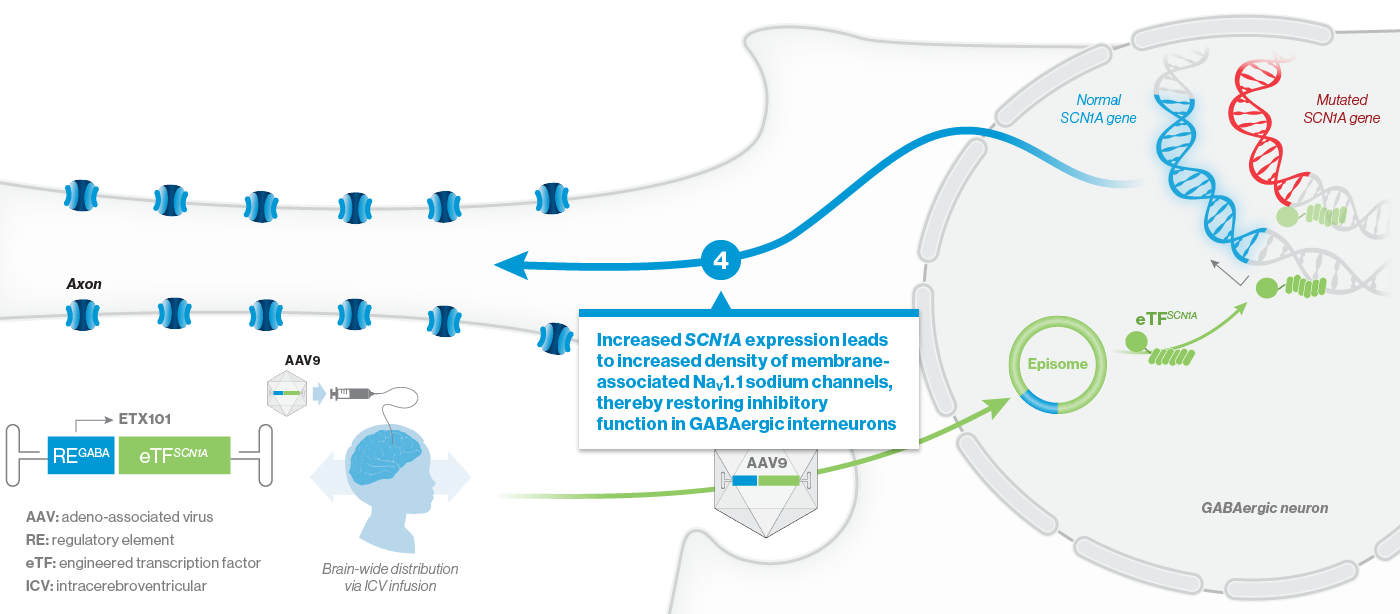
NaV1.1 is primarily expressed in inhibitory interneurons, the cell type responsible for reducing neuronal activity in the brain. With reduced NaV1.1, the brain may become hyperactive, causing seizures and other clinical manifestations that significantly impact the lives of affected children and their families. Currently approved Dravet syndrome treatments are chronically administered and purely symptomatic, aiming only to reduce seizure frequency or severity. Despite the currently available treatments, very few people with Dravet syndrome experience sustained periods of seizure freedom.
Sources: Wu YW, et al. Pediatrics 2015;136:e1310–15; www.dravetfoundation.org; NORD; Symonds JD, et al. Hum Mutat. 2020 Feb;41(2):363-374; Gataullina S, Dulac O. Seizure 2017;44:58–64; Catarino CB, et al. Brain 2011;134:2982–3010; Wirrell EC, et al. Pediatr Neurol 2017;68:18–34.e3; Lagae L, et al. Dev Med Child Neurol 2018;60:63–72; Skluzacek JV. Epilepsia. 2011;52(Suppl 2):95-101; Cooper MS. Epilepsy Res. 2016;128:43-47

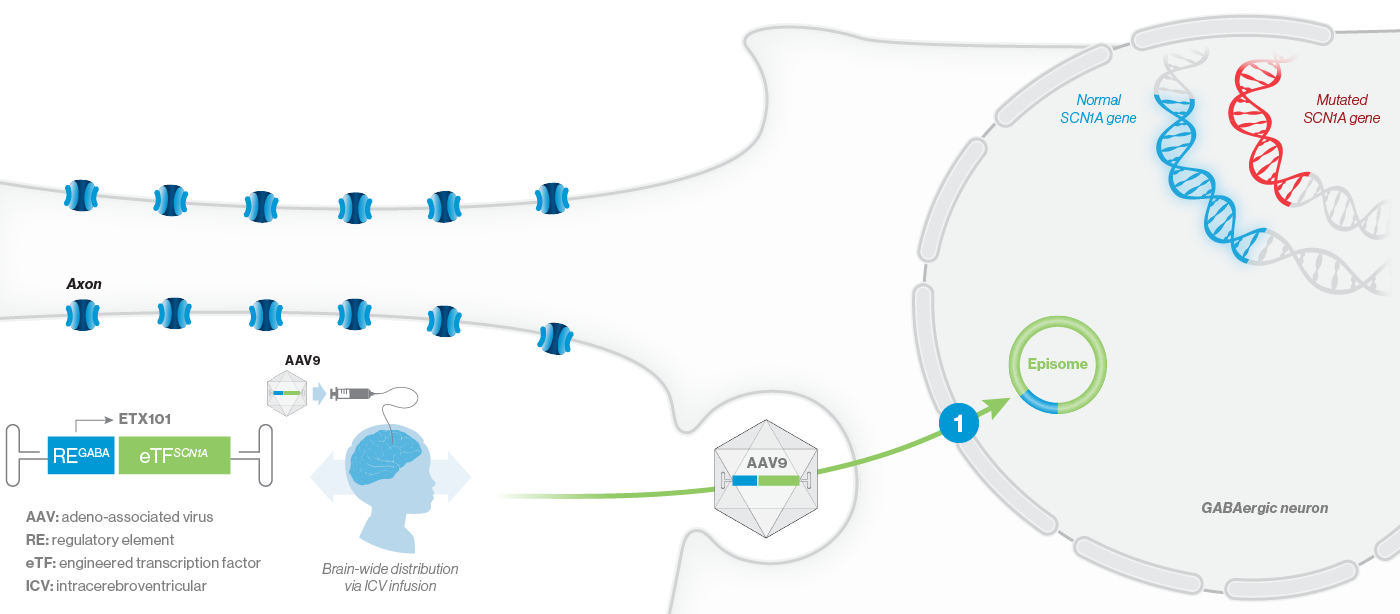
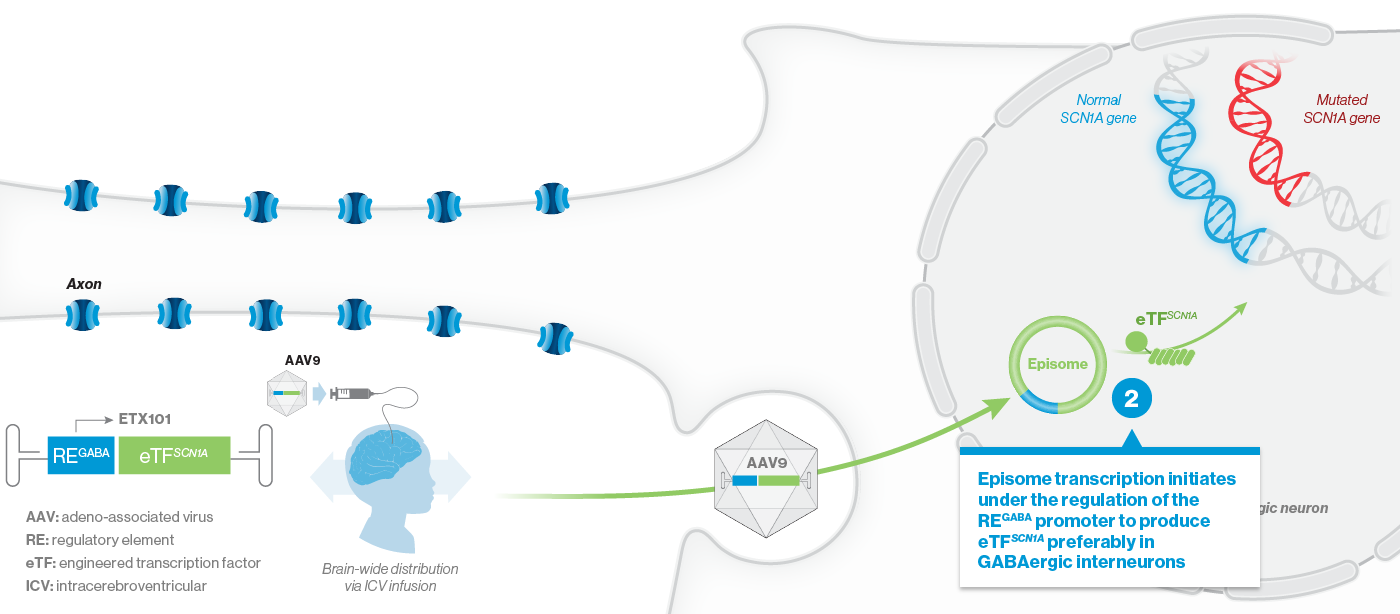
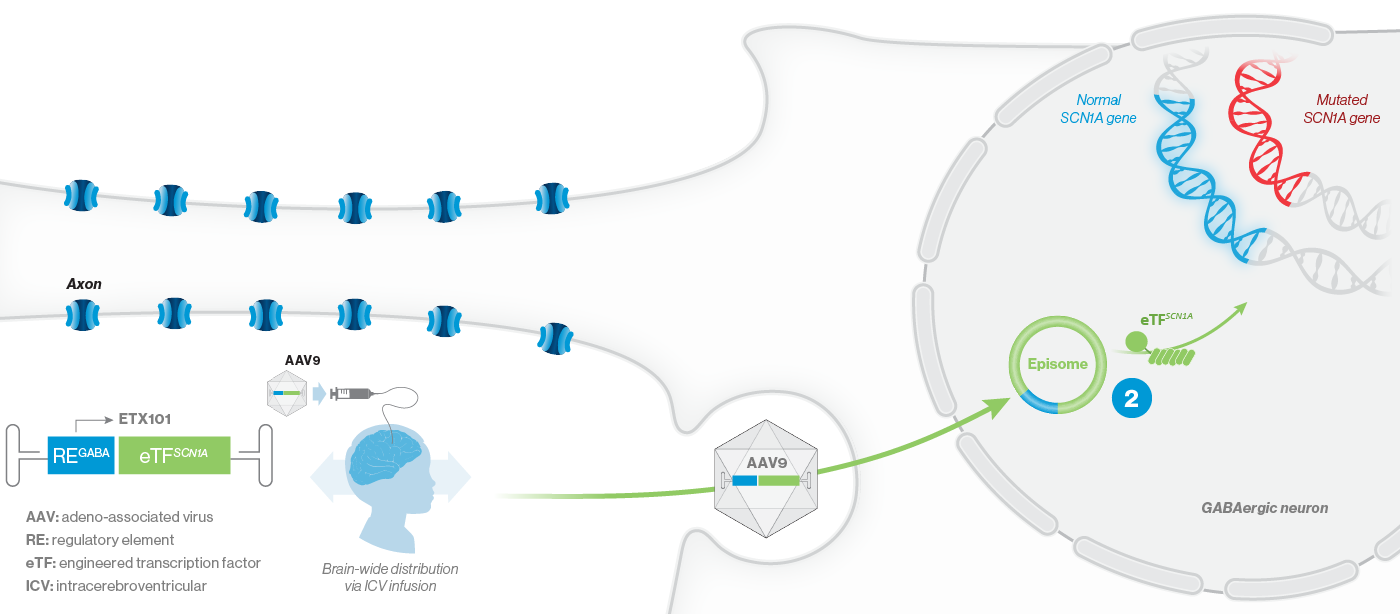
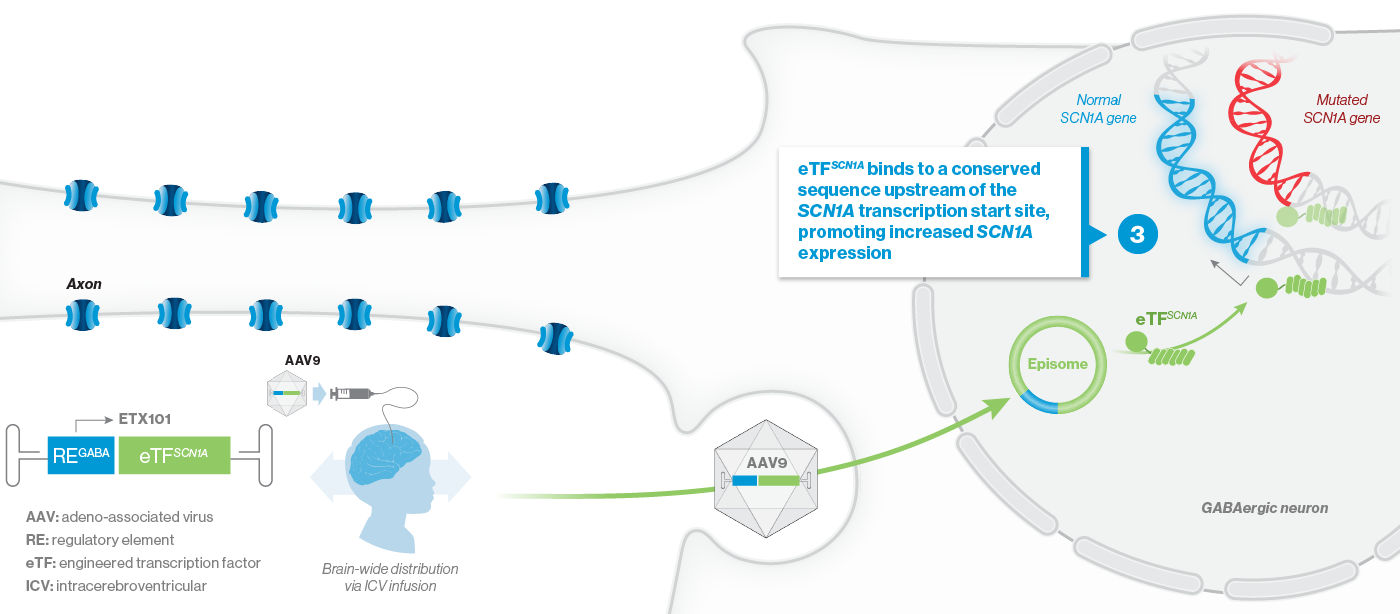
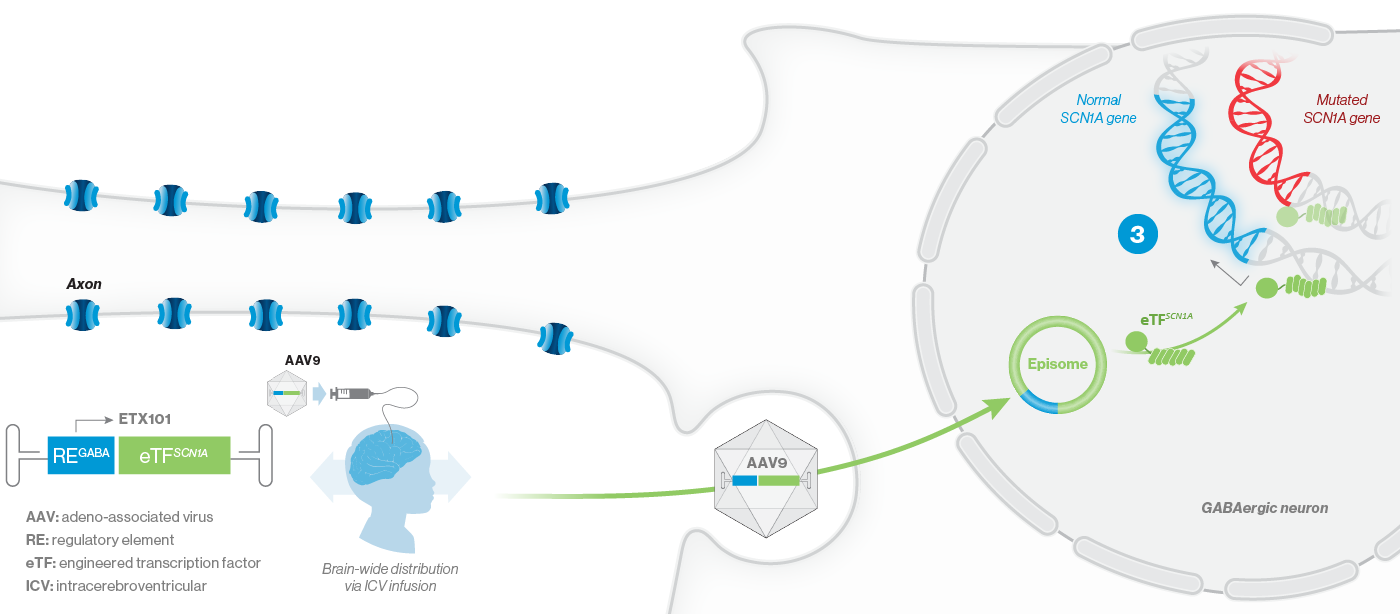
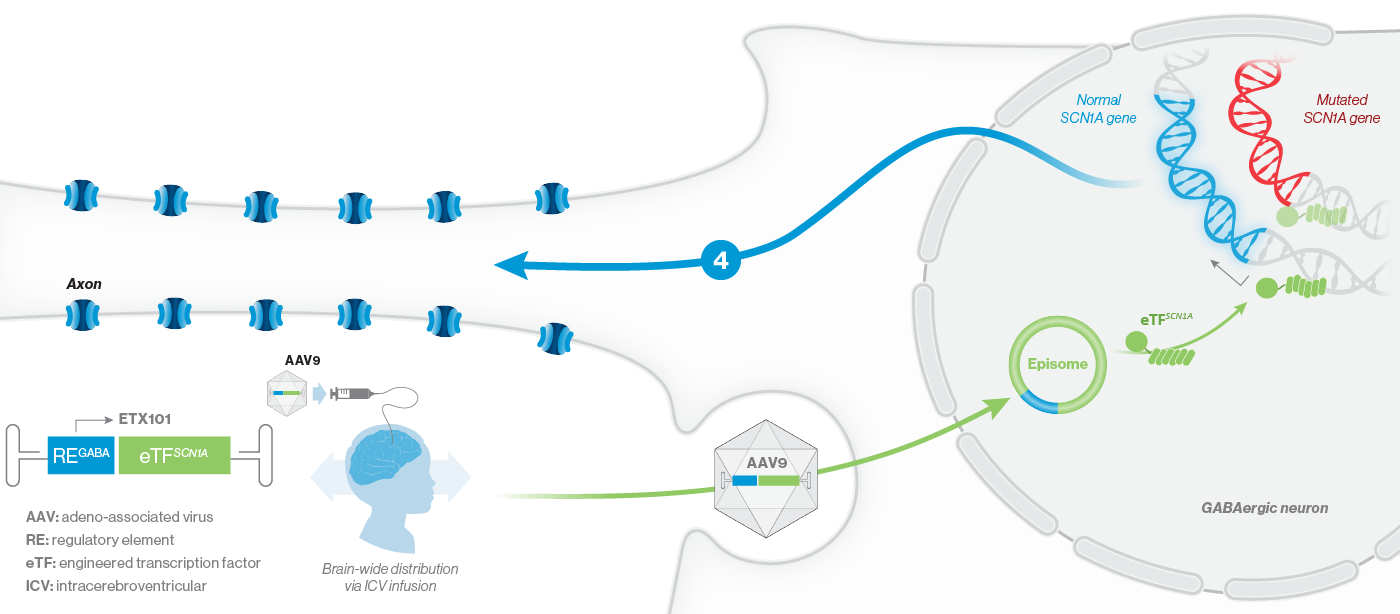
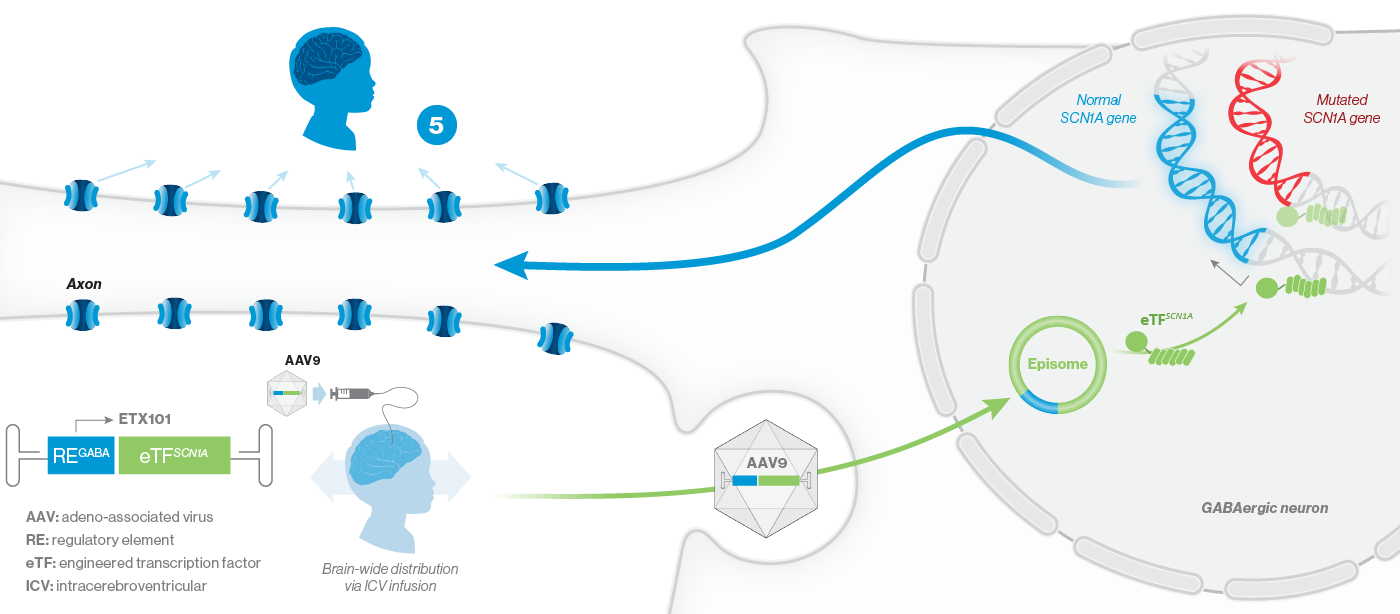
How ETX101 is Expected to Work
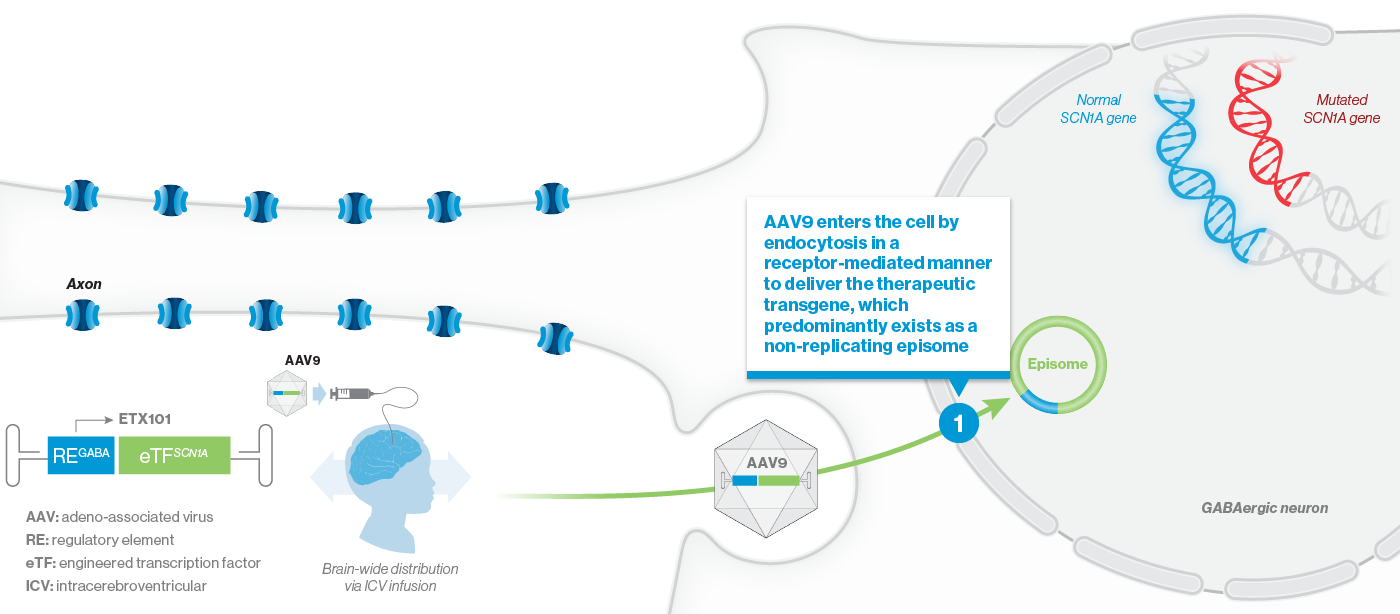
ETX101 utilizes a clinically validated adeno-associated virus (AAV) capsid that has been extensively used in clinical trials for CNS disorders, as well as in a product with regulatory approval to treat spinal muscular atrophy, a rare neuromuscular disease. ETX101 is delivered via an intracerebroventricular (ICV) infusion, which is considered by neurosurgeons to be a safe and standard procedure to deliver antibiotics and enzyme-replacement therapies, among other drugs. Local ICV administration of AAV-mediated gene therapy to the CNS enhances delivery of the drug to key brain structures important for seizures and neurocognition, thereby increasing the potential for efficacy with a one-time administration.
Our preclinical data package for ETX101 demonstrates broad distribution in relevant CNS structures in non-human primates (NHPs), as well as long-term survival and reduction of seizures in the Dravet mouse model.
Underlying Cause of Dravet
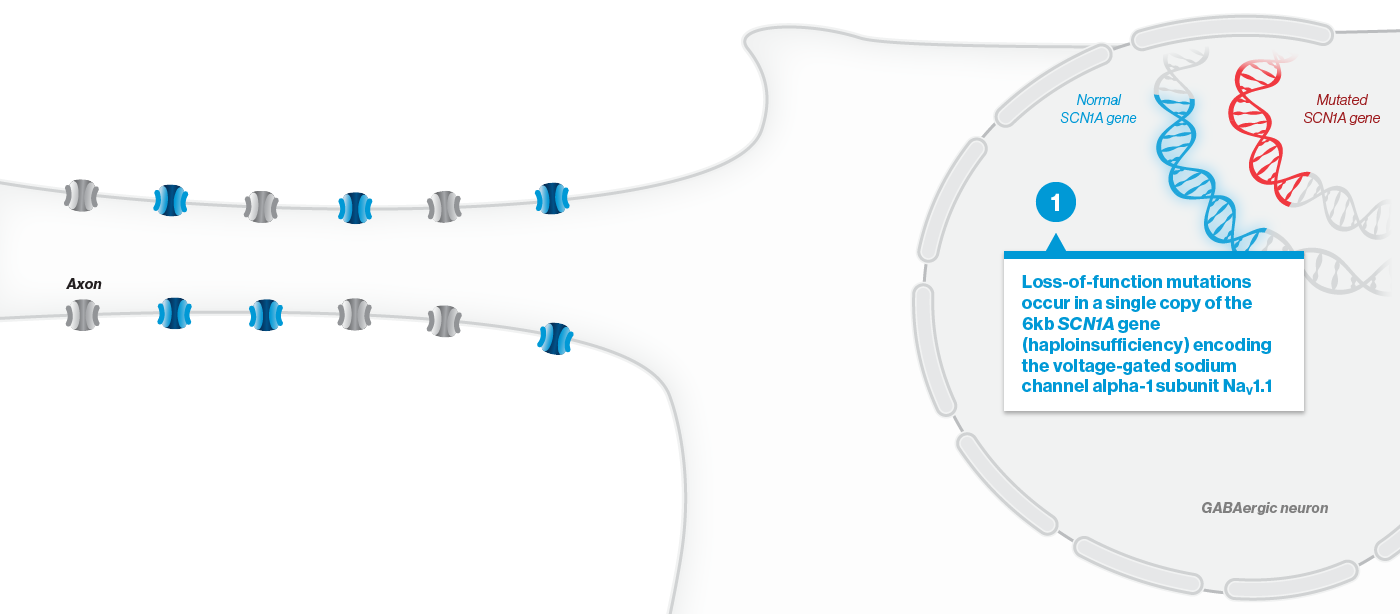
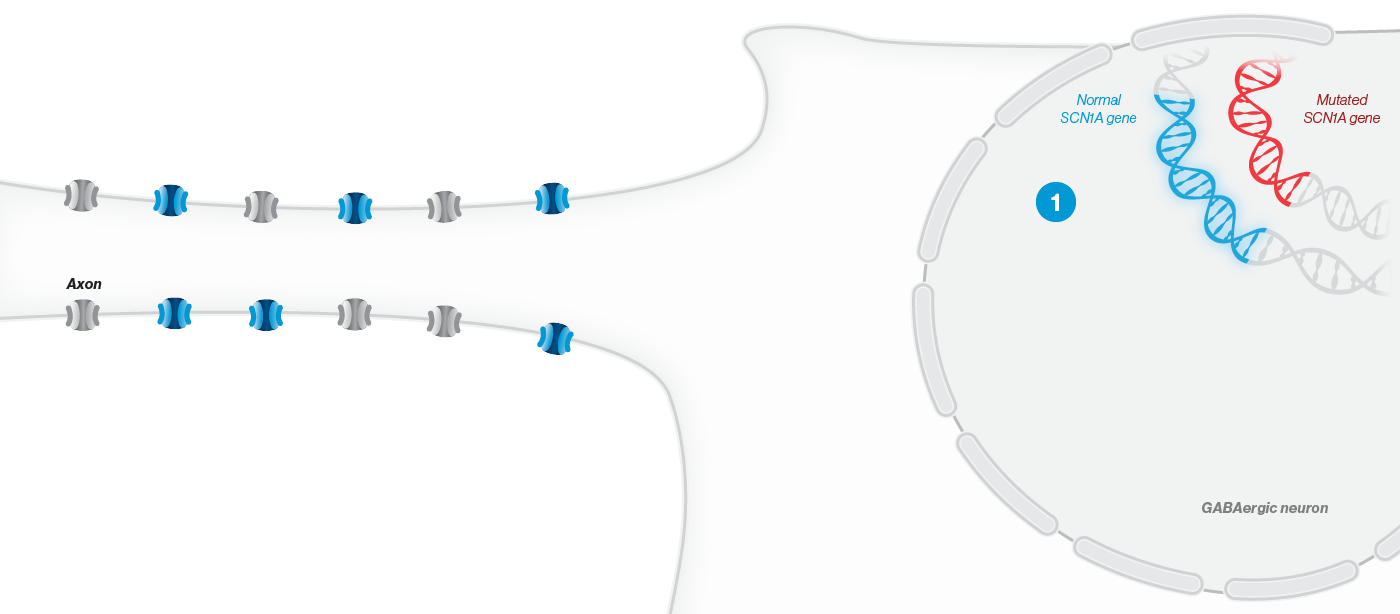
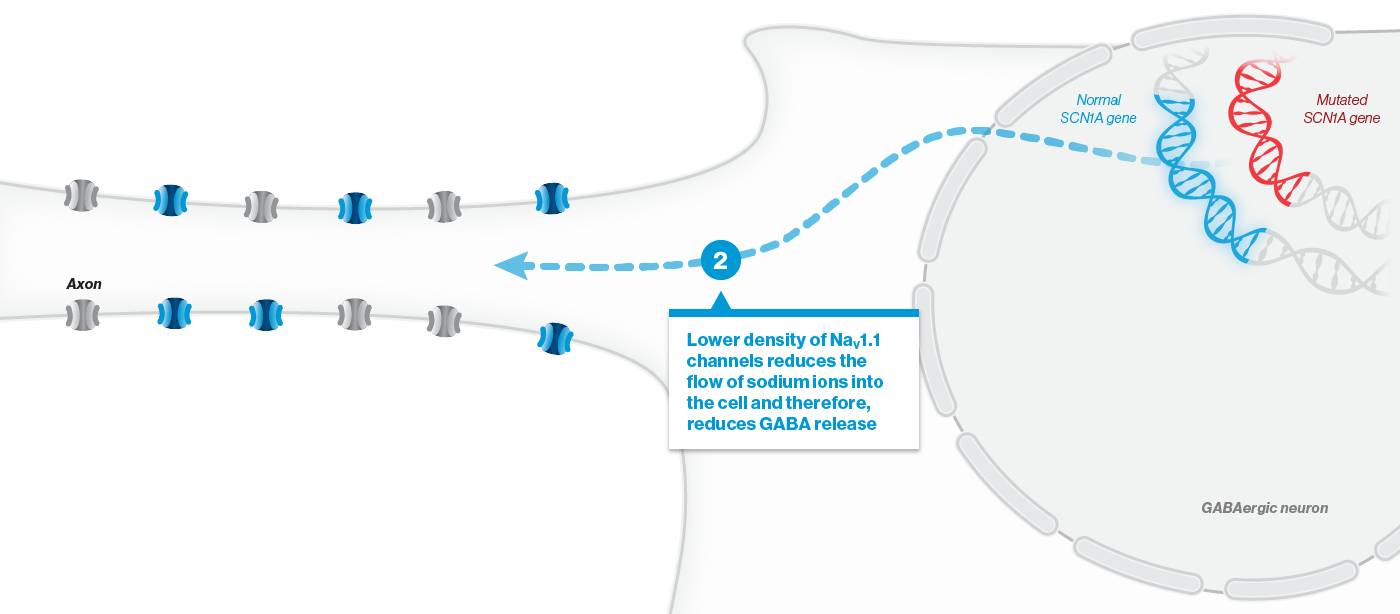
How ETX101 is Expected to Work
Developing ETX101
ETX101 has been generated using Encoded’s in-house capabilities – from preclinical discovery to initiation of clinical trials. We anticipate leveraging this infrastructure for future development programs, paving an accelerated path from basic science through to the clinic.
